Tau Deposition in Alzheimer’s Disease: Why Tau Predicts Symptoms Better Than Amyloid
 If amyloid explains why Alzheimer’s disease begins, tau explains why it progresses, where it progresses, and how severe it becomes. Over the last decade, converging evidence from neuropathology, tau-PET imaging, and longitudinal clinical studies has firmly established tau deposition as the strongest biological predictor of symptom severity and outcome in Alzheimer’s disease (AD).
If amyloid explains why Alzheimer’s disease begins, tau explains why it progresses, where it progresses, and how severe it becomes. Over the last decade, converging evidence from neuropathology, tau-PET imaging, and longitudinal clinical studies has firmly established tau deposition as the strongest biological predictor of symptom severity and outcome in Alzheimer’s disease (AD).
This article examines how tau pathology maps onto cognition, behavior, disease trajectory, and prognosis—using the most current mechanistic and clinical understanding.
Tau: from stabilizer to disruptor
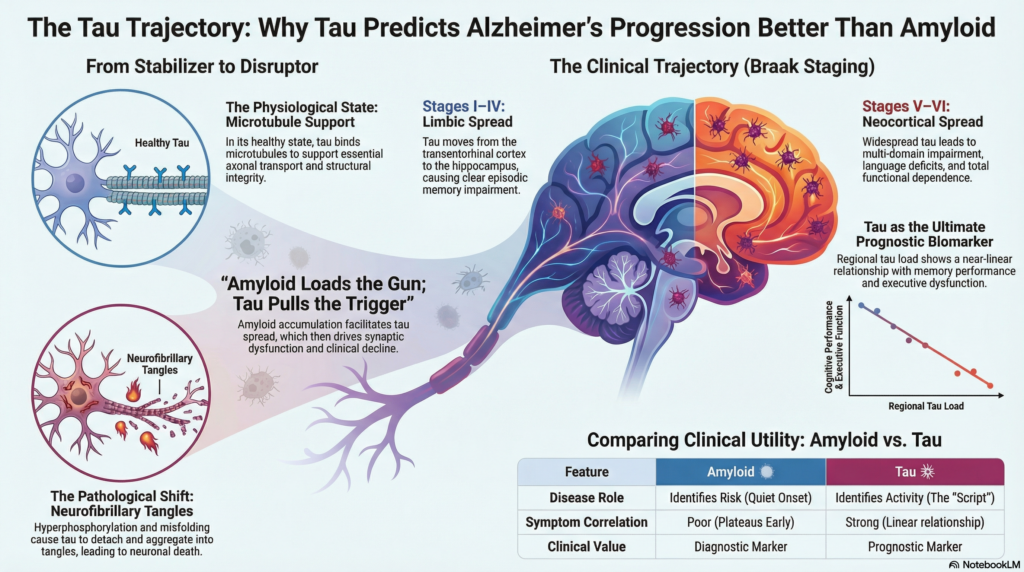
Tau is a microtubule-associated protein essential for axonal transport and neuronal structural integrity. In its physiological state, tau binds microtubules and supports efficient intracellular trafficking.
In Alzheimer’s disease, tau undergoes:
-
Hyperphosphorylation
-
Misfolding and truncation
-
Detachment from microtubules
This destabilizes axonal transport and leads to aggregation into paired helical filaments, forming neurofibrillary tangles (NFTs)—the pathological hallmark most closely linked to neuronal dysfunction and death.
Braak staging: tau follows symptoms, not time
Classic Braak and Braak staging, now validated by tau-PET imaging, demonstrates a striking clinico-anatomical relationship.
-
Stages I–II (Transentorhinal / Entorhinal cortex)
Minimal or no symptoms. Subtle episodic memory inefficiency may be detectable only on sensitive testing. -
Stages III–IV (Limbic spread – hippocampus, amygdala)
Clear episodic memory impairment, impaired learning, and early executive dysfunction. -
Stages V–VI (Neocortical spread)
Multidomain cognitive impairment, language deficits, visuospatial dysfunction, behavioral changes, and functional dependence.
The critical insight is this: tau distribution mirrors clinical phenotype far more precisely than amyloid burden.
Tau burden correlates with cognitive decline
Multiple longitudinal studies now show that:
-
Regional tau load correlates strongly with current cognitive impairment
-
Baseline tau predicts rate of future decline
-
Tau progression predicts conversion from MCI to dementia
In contrast, amyloid burden plateaus early and correlates poorly with symptom severity once dementia begins.
Tau-PET uptake in the temporal and parietal cortices shows near-linear relationships with:
-
Memory performance
-
Executive dysfunction
-
Language deficits
-
Global cognitive scales (MMSE, MoCA, CDR-SB)
Simply put: amyloid loads the gun; tau pulls the trigger.
Tau spread is network-driven
Modern network neuroscience has reframed tau propagation as a trans-synaptic process:
-
Tau spreads along functionally connected brain networks
-
High-connectivity hubs (default mode network) are especially vulnerable
-
Synaptic activity accelerates tau release and uptake
This explains why tau deposition follows cognitive networks rather than vascular territories—and why clinical phenotypes differ despite similar amyloid loads.
Interaction between amyloid and tau
Amyloid and tau are not independent processes.
Current evidence supports a two-stage model:
-
Amyloid accumulation facilitates tau misfolding and spread
-
Tau drives synaptic dysfunction, neurodegeneration, and clinical decline
Without amyloid, tau pathology often remains localized and indolent (as seen in primary age-related tauopathy, PART). Amyloid acts as a biological accelerator for tau toxicity.
Tau predicts outcome and prognosis
Tau burden is now recognized as a predictor of:
-
Speed of cognitive decline
-
Functional disability
-
Institutionalization risk
-
Mortality
Patients with high neocortical tau load show:
-
Faster decline despite similar amyloid levels
-
Poorer response to symptomatic treatments
-
Greater likelihood of neuropsychiatric symptoms (apathy, agitation)
Tau is therefore a prognostic biomarker, not merely a diagnostic one.
Clinical implications: why tau matters more in practice
This shift has profound implications.
-
Early amyloid positivity identifies risk
-
Tau positivity identifies disease activity
In real-world clinical settings, tau burden helps explain:
-
Why some amyloid-positive individuals remain cognitively intact
-
Why others decline rapidly despite modest amyloid load
-
Why anti-amyloid therapies show limited benefit once tau is widespread
Emerging anti-tau therapies, synaptic stabilizers, and network-level interventions are increasingly viewed as essential complements—or successors—to amyloid-focused strategies.
Reframing Alzheimer’s disease
Alzheimer’s disease is not defined by what accumulates first, but by what destroys neurons last.
Amyloid sets the stage quietly and early.
Tau determines the script, the pacing, and the ending.
Understanding tau deposition allows clinicians to move beyond binary diagnoses toward trajectory-based prognostication, offering patients and families more realistic expectations and better-timed interventions.
About the Author
Dr. Srinivas Rajkumar T, MD (AIIMS, New Delhi), DNB, MBA (BITS Pilani)
Consultant Psychiatrist
Dr. Srinivas Rajkumar has a focused clinical and academic interest in amyloid–tau biology, Alzheimer’s disease progression, and early cognitive disorders. His work emphasizes translating advances in biomarker science—amyloid, tau, sleep, and vascular factors—into clinically meaningful risk stratification and management.
He practices at Mind & Memory Clinic, Apollo Clinic, Velachery, Chennai, where he evaluates memory complaints, mild cognitive impairment, and dementia spectrum disorders, with a strong emphasis on early detection, prognostication, and evidence-based counseling.
📍 Apollo Clinic, Velachery, Chennai
📞 +91-8595155808
✉️ srinivasaiims@gmail.com